Research/ Bioinformatics
Introduction
Our laboratory primarily conducts image processing research, but we also pursue topics beyond imaging. One such area is bioinformatics—the fusion of life science and information science, well known for applications such as genome analysis. Within this field, we design artificial nucleic acids called aptamers. Aptamers are nucleotide sequences that bind to specific proteins and are applied to biopharmaceuticals and molecular sensing. Here we describe research on discovering aptamers from vast nucleic acid sequence pools.
Aptamers
An aptamer is an artificially created nucleic acid sequence designed to bind to a specific protein.
Aptamer Selection via HT-SELEX
Because the nucleotide sequence that binds to a given target molecule is unknown, it must be discovered experimentally from a large pool of nucleic acids. The standard approach is Systematic Evolution of Ligands by EXponential enrichment (SELEX). Recently, HT-SELEX combined with next-generation sequencing (NGS) has been widely adopted. The six steps of HT-SELEX are as follows.
- Synthesize an initial library comprising randomly initialized nucleic acid molecules.
- Mix with the target molecule to allow binding between nucleic acids and the target.
- Wash away unbound nucleic acids, retaining only those that bound.
- Elute bound nucleic acids from the target molecule.
- Amplify eluted nucleic acids by polymerase chain reaction (PCR).
- Read sequence data with a next-generation sequencer.
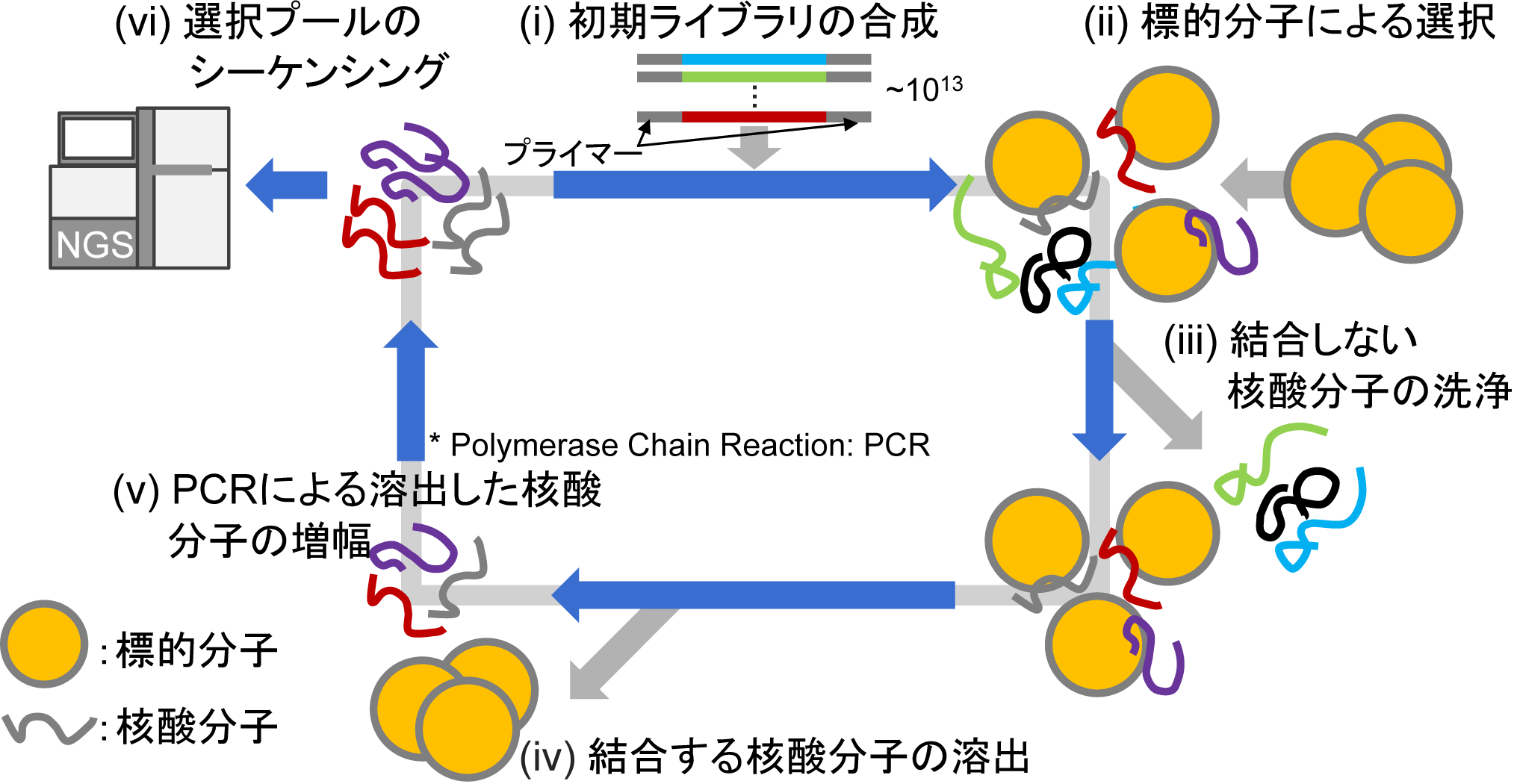
Figure 1: Aptamer selection via HT-SELEX
Clustering of Sequence Data
To find aptamers from massive sequence data obtained by HT-SELEX, one would ideally evaluate every sequence experimentally—but this is impractical in cost and time. Computational identification of sequences with high affinity to the target would greatly reduce both. Our laboratory has proposed clustering methods to support aptamer selection. For details, see reference [1].
Summary
We have briefly introduced our bioinformatics research. This work is conducted jointly with the Bio Lab at NEC Solution Innovators, Ltd.